Members involved: Denis Jacquemin (PR), Adèle Laurent (CR), Morgane Vacher (CR), Smida Hassiba (ATER), Pauline Vérité (ANR PhD), et Amara Charyteh (LUMOMAT PhD)
General presentation
Within the framework of this thematic, the objective is to simulate the properties linked to electronic excited states with a view to, not only, interpret the experimental data but also to predict and optimize the characteristics of new compounds. The state of the art is used to simulate the absorption and emission spectra of compounds in the condensed phase or in a heterogeneous complex environment (DNA, protein, cage, etc.).
The theoretical tools allowing us to carry out this work are very diverse, ranging from quantum methods (QM) most often based on electronic density (TD-DFT, BSE) or based on wave function (CIS, CC2, ADC (2 ), CCSD, CC3…) to hybrid methods combining quantum mechanics and molecular mechanics (QM / MM). The team also simulates the non-adiabatic dynamics of the molecular system in a semi-classical (surface hopping, Ehrenfest) or quantum (DD-vMCG) manner in order to obtain a complete and intuitive image of the electronic states and nuclear structures visited over time after excitation. Four axes are thus defined (i) dyes, (ii) photochromes, (iii) biological systems and (iv) attosecond science.
Some recent reviews:
- F. Loos, D. Jacquemin, ChemPhotoChem. 2019, in press
- Jacquemin, I. Duchemin, X. Blase Chem. Soc. Rev. 2018, 47, 1022-1043.
- Vacher, M. J. Bearpark, M. A. Robb, Direct methods for non-adiabatic dynamics: connecting the single-set variational multi-configuration Gaussian (vMCG) and Ehrenfest perspectives, Theor. Chem. Acc. 2016, 135, 187.
- Escudero, A. D. Laurent, D. Jacquemin, Time-Dependent Density Functional Theory: A Tool to Explore Excited States, Handbook of Computational Chemistry (2016), page 1-35.
- Fihey, A. Perrier, W. R. Browne, D. Jacquemin, Chem. Soc. Rev. 2015, 44, 3719-3759
- D. Laurent, C. Adamo, D. Jacquemin, Phys. Chem. Chem. Phys. (perspective) 2014, 16, 14334–56
- Adamo, D. Jacquemin, Chem. Soc. Rev. 2013, 42, 845–56
- D Laurent, Denis Jacquemin, TD-DFT benchmarks: A review, Int. J. Quantum Chem. 2013, 113, 2019–2039
Organic dyes are a family of compounds of significant industrial interest for "green" applications. Indeed, most of recent efforts have been focused on clean technologies such as photovoltaic cells. The most sought dyes are those which absorb in the infrared, which have a large quantum yield, a great (photo) stability, etc. In this context, the group has the technology to model the absorption and emission of these molecules in condensed phase taking into account the vibronic coupling.
The methodology used allows the experimental data to be reproduced and explained. It is therefore possible to predict variations in optical spectra (shapes and positions) during auxochromic substitutions, variations in pH, effects of solvochromism and acidochromism ... and therefore to propose new effective dyes to experimenters.
Numerous varieties of dyes have been studied in this thematic, therefore only recent subjects are highlighted here. This axis notably includes (i) the study of cyanine derivatives (BODIPY, Aza-BODIPY, BORANIL, ..) which constitutes a theoretical challenge, (ii) understanding and characterizing the reactivity of the derivatives of original macrocyclics comprising zwitterions and (iii) simulating the properties of dyes exhibiting an excited state intramolecular proton transfer (ESIPT).
Note that the test bench studies (choice of the functional in DFT, the basis of atomic functions, the solvent model, the method, etc.) making it possible to define a protocol in order to characterize the excited states (vibronic spectra and vertical transitions) are found in the thematic methodological developments (Ax-2).
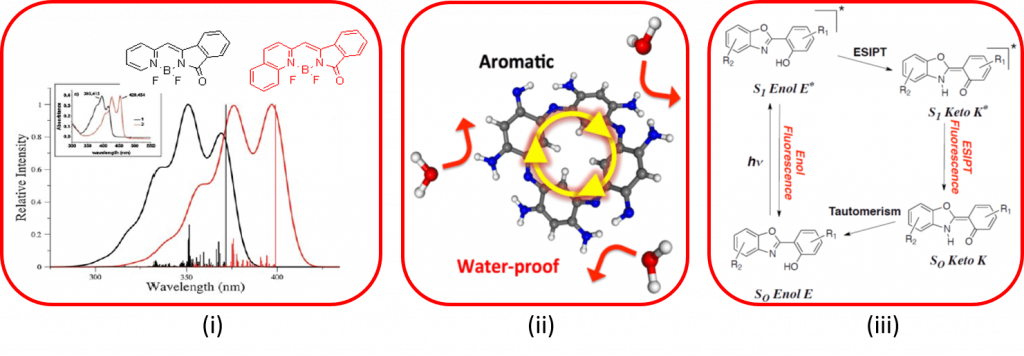
Significant articles :
- A.M. Grabarz, et al., J. Org. Chem. 2016, 81, 2280–2292. S. Chibani et al., Chem. Sci. 2013, 4, 1950-63. A.M. Grabarz, et al., Dyes Pigm. 2019, 170, 170481.
- G. Marchand, et al., Phys. Chem. Chem. Phys. 2016, 18, 9608-9615. S. Pascal, et al, Mater Chem. Front 2018, 2, 1618-1625. S. Pascal, et al, J. Org. Chem. 2019, 84, 1387-1397.
- Simon Budzak, Denis Jacquemin, J. Phys. Chem.B, 2016, 120, 6730-6738. C. Azarias, et al., Chem. Sci. 2016, 7, 3763-3774. K. Benelhadj et al., Chem. Eur. J. 2014, 20, 1-16. P. Vérité, C. A. Guido, D. Jacquemin, Phys. Chem. Chem. Phys. 2019, 21, 2307-2317. P. Vérité, S. Hédé, D. Jacquemin, Phys. Chem. Chem. Phys. 2019, 21,17400-17409.
A photochrome is a molecule which can appear in two forms (closed / open, cis / trans, ...), the passage from one form to another being induced thermally or photochemically via the absorption of energy radiations or of different wavelengths (UV, visible or infrared). This photochromic reaction is reversible between the two molecular states which have very different electronic and therefore optical properties. Organic photochromic molecules have undeniable potentials for their use in the transmission and storage of a data bit (0 or 1) by optical or electronic means.
Our objectives in this area are (i) to improve photochromic compounds by maximizing the linear and non-linear optical properties of the two electronic states, (ii) to simulate the electronic properties of multi-photochromes (molecular entity comprising two or several photochromes) allowing to increase the quantity of stored data and (iii) to model the interactions and the electronic properties between a (multi-) photochromic and a semiconductor surface (NiO, TiO2, Cu,…). These works being carried out with (TD-) DFT or multi-reference methods and including the periodic boundary conditions for the surface calculations.
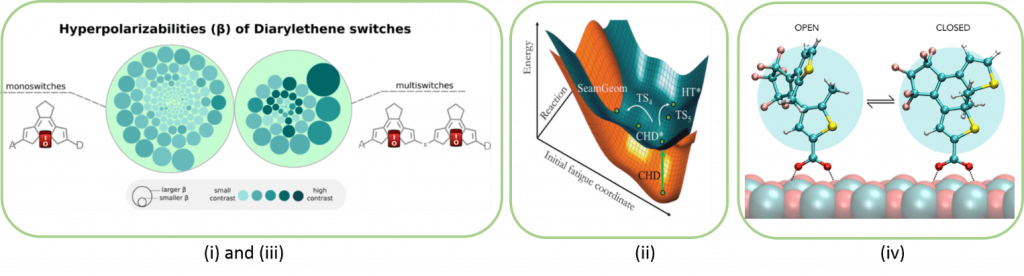
Reference publications:
- K. J. Chen et al., J. Phys Chem C 2014, 118, 4334-45. T. Jaunet-Lahary et al., J. Phys Chem C 2014, 18, 28831-84
- D. Mendive-Tapia et al., Phys. Chem. Chem. Phys. 2014, 16, 18463–71
- A. Fihey, D. Jacquemin Chem. Sci. 2015, 6, 3495-3503. A. Fihey, A. Perrier, W. R. Browne, D. Jacquemin, Chem. Soc. Rev. 2015, 44, 3719-3759. B. Lasorne et al. Chem. Sci. 2015 6, 5695-5702. A. Perrier, F. Maurel, D. Jacquemin Acc. Chem. Res. 2012, 45, 1173–82
- K. J Chen et al. J. Mater. Chem. A 2016, 4, 2217-2227. K. J. Chen, F. Boucher, D. Jacquemin J. Phys. Chem. C 2015, 119, 16860-16869. K. J. Chen et al., J. Phys Chem C 2015, 119, 3684-3696. A. Fihey, B. Le Guennic, D. Jacquemin, J. Phys. Chem. Lett. 2015, 6, 3067-3073.
This research axis involves the modeling of macromolecules which are naturally or artificially fluorescent using complementary approaches combining virtual screening, molecular modeling and QM / MM hybrid methods in order to characterize their spectral properties. These two categories of fluorescent macromolecules are widely used in medical imaging but also in biology for the detection of diseases or anomalies.
Naturally fluorescent macromolecules intrinsically include a chromophore, while artificial ones are modified by site-directed mutagenesis by introducing a molecular probe (or marker) in place of an amino acid into a protein of interest. The spectral modifications induced by the presence, or not, of a ligand / drug, make it possible to obtain information very locally about the position of the ligand.
The objectives of this axis are (i) to extend the color palette of new naturally fluorescent proteins by proposing new chromophores or new mutations within the photoactive site and (ii) to determine in proteins of therapeutic interest the position of a ligand or drug in order to be able to modulate its activity, in close collaboration with groups of experimenters.
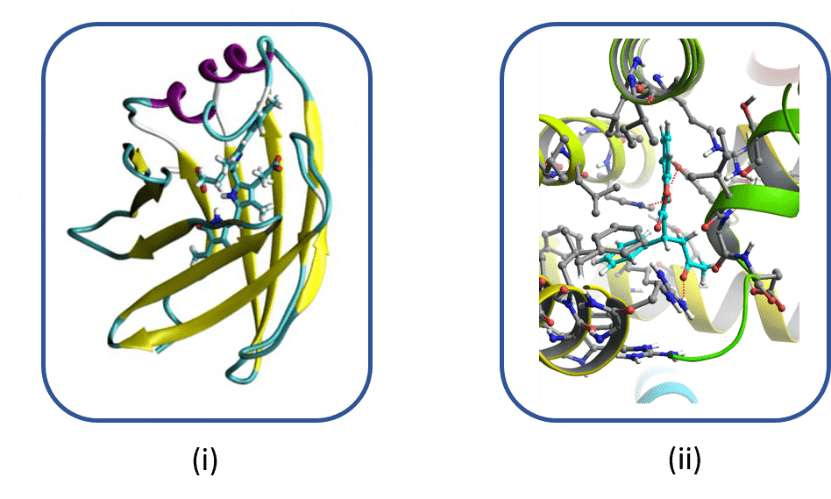
In order to understand photochemical reactions, experiments resolved in time with a temporal resolution of the order of a femtosecond (1 fs = 10-15 s) played a major role in probing the movement of the nuclei in real time. Recent advances in attosecond science (1 as = 10-18 s) open up the possibility of observing and controlling the movement of electrons on their intrinsic time scale. Because of the time-energy uncertainty principle, these extremely short duration pulses have a large spectral width.
They can therefore be used to populate several excited electronic states in a coherent manner: this is called an electronic wave packet. Such a wave packet has a new electronic distribution (which is not the simple average of the electronic distributions of the different states). It can therefore be considered as a new type of initial electronic state.
This research axis aims to deepen the understanding of the processes of coherent electronic dynamics and to study their application to photochemical reactions. This includes in particular (i) studying the process of electronic decoherence and (ii) studying the nuclear dynamics induced by an electronic wave packet.
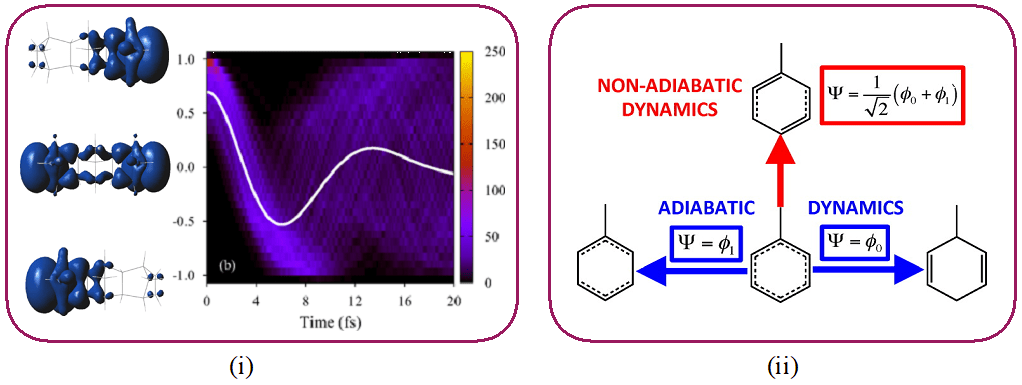
Reference publications:
- M. Vacher, M. J. Bearpark, M. A. Robb, J. P. Malhado, Phys. Rev. Lett. 2017, 118, 083001. M. Vacher, L. Steinberg, A. J. Jenkins, M. J. Bearpark, M. A. Robb, Phys. Rev. A Rapid Communication 2015, 92, 040502(R). A. J. Jenkins, M. Vacher, R. M. Twidale, M. J. Bearpark, M. A. Robb, J. Chem. Phys. 2016, 145, 164103.
- M. Vacher, F. E. Albertani, A. J. Jenkins, I. Polyak, M. J. Bearpark, M. A. Robb, Faraday Discuss. 2016, 194, 95-115. M. Vacher, J. Meisner, D. Mendive-Tapia, M. J. Bearpark, M. A. Robb, J. Phys. Chem. A, 2015, 119, 5165-5172.
Collaborations:
- Carlo Adamo, Ilaria Ciofini et Aurélie Perrier (Paris Tech) Institut de recherche Chimie Paris, équipe de Modélisation des Systèmes Complexes (link)
- Benedetta Mennucci (université de Pise) DCCI “Dipartimento di Chimica e Chimica Industriale” Mennucci Research Group (link)
- Boris Le Guennic (iSCR Rennes « Institut des sciences chimiques de Rennes ») UMR 6226, Chimie théorique inorganique groupe) (link)
- Pierre-François Loos (LCPQ, UMR 5626, Université de Toulouse, Toulouse) (link)
- Xavier Blase (Institut Néel, université de Grenoble-Alpes) équipe de Matière condensée théorique (link)
- Olivier Siri, Simon Pascal (Université Aix-Marseilles) Laboratoire CiNAM, UMR 7325 Ingénierie moléculaire et matériaux fonctionnels (link)
- Gilles Ulrich (ICPEES – Institut de Chimie et Procédés pour l’Énergie, l’Environnement et la Santé) (link)
- Miroslav Medved, Simon Budzak (Matej Bel University, Banska Bystrica Slovaquie) (link)