Overview
These research activities are focused on the development of mono- and multivalent glyco-conjugates to modulate the activity of lectins and glycosidases of pathogens, the coating of viral particles for gene therapy, and the development of new bioconjugation methods.

Research
Selected articles
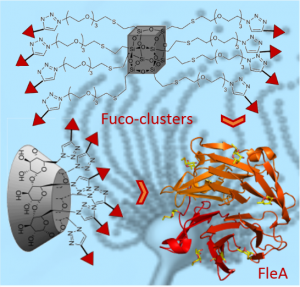
1 - Heptylmannose-functionalized cellulose for the binding and specific detection of pathogenic E.coli Madeleine Cauwel, Adeline Sivignon, Clarisse Bridot, Medy C. Nongbe, David Deniaud, Benoit Roubinet, Ludovic Landemarre, François-Xavier Felpin, Julie Bouckaert, Nicolas Barnich, Sébastien G. Gouin.* Chem. Commun 2019, 55 (68), 10158-10161. We developed a chemical method to covalently functionalizecellulose nanofibers and cellulose paper with mannoside ligands displaying a strong affinity for the FimH adhesin from pathogenic E. coli strains. Mannose-grafted cellulose proved efficient to selectively bind FimH lectin and discriminate pathogenic E. coli strains from non-pathogenic ones. These modified papers are valuable tools for diagnosing infections promoted by E.coli, such as cystitis or inflammatory bowel diseases, and the concept may be applicable to other lifethreatening pathogens. 2 - Multivalent fucosides with nanomolar affinity for the Aspergillus fumigatus lectin FleA prevent spore adhesion to pneumocytes
Victor Lehot, Yoan Brissonnet, Christophe Dussouy, Sami Brument, Aurore Cabanettes, Emilie Gillon, David Deniaud, Annabelle Varrot, Patrice Le Pape, and Sébastien G. Gouin*,Chem. Eur. J. 2018, 24, 19243-19249.
The FleA receptor in the opportunistic pathogen Aspergillus fumigatus was shown to be involved in spore (conidium) adherence to airway mucins and bronchoepithelial cells. We developed multivalent fucosides with various valencies and spacer arm lengths as conidium anti-adhesives. The hexa- and octavalent fucosides were nanomolar antagonists of FleA, strongly inhibiting conidium adherence to pneumocytes.
2 - Multivalent fucosides with nanomolar affinity for the Aspergillus fumigatus lectin FleA prevent spore adhesion to pneumocytes
Victor Lehot, Yoan Brissonnet, Christophe Dussouy, Sami Brument, Aurore Cabanettes, Emilie Gillon, David Deniaud, Annabelle Varrot, Patrice Le Pape, and Sébastien G. Gouin*,Chem. Eur. J. 2018, 24, 19243-19249.
The FleA receptor in the opportunistic pathogen Aspergillus fumigatus was shown to be involved in spore (conidium) adherence to airway mucins and bronchoepithelial cells. We developed multivalent fucosides with various valencies and spacer arm lengths as conidium anti-adhesives. The hexa- and octavalent fucosides were nanomolar antagonists of FleA, strongly inhibiting conidium adherence to pneumocytes.
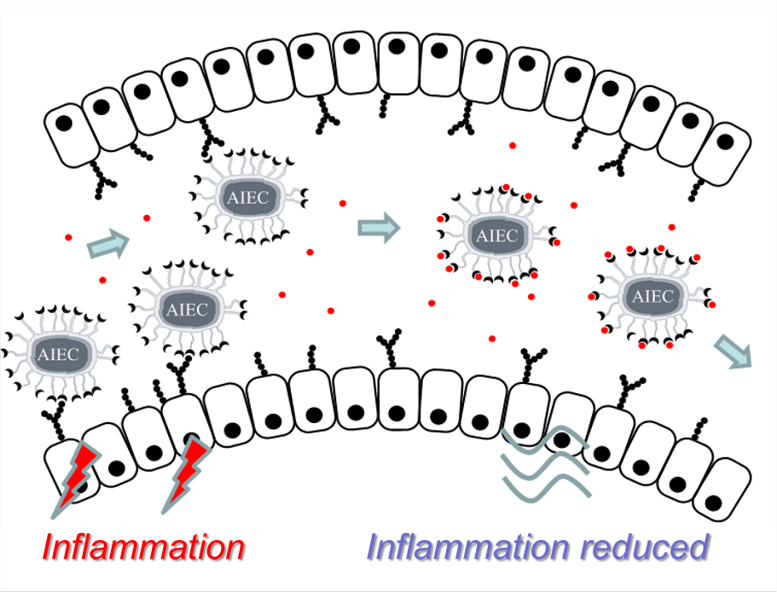
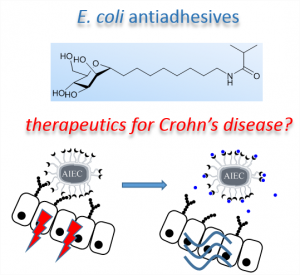
 3 - The anti-adhesive strategy in crohn’s disease: orally active mannosides to decolonize pathogenic Escherichia coli from the gut.
Alvarez Dorta, D.; Sivignon, A.; Chalopin, T.; Dumych,T.; Pecina, A.; Roos, G.; Bilyy, R.; Deniaud, D.; de Ruyck, J.; Bouckaert, J.; Barnich, N.; Gouin S. G. ChemBioChem, 2016, 17, 1-18.
Ready for take off? Adherent-invasive E. coli (AIEC) were shown to promote the inflammation of the intestinal mucosa of patients with Crohn’s disease (CD). In this work, we designed a small library of AIEC antiadhesives. A lead compound was shown to decolonize AIEC from the gut of a transgenic mouse model of CD after oral administration. The water-soluble and non-cytotoxic compounds developed may open new perspectives in the treatment of CD.
3 - The anti-adhesive strategy in crohn’s disease: orally active mannosides to decolonize pathogenic Escherichia coli from the gut.
Alvarez Dorta, D.; Sivignon, A.; Chalopin, T.; Dumych,T.; Pecina, A.; Roos, G.; Bilyy, R.; Deniaud, D.; de Ruyck, J.; Bouckaert, J.; Barnich, N.; Gouin S. G. ChemBioChem, 2016, 17, 1-18.
Ready for take off? Adherent-invasive E. coli (AIEC) were shown to promote the inflammation of the intestinal mucosa of patients with Crohn’s disease (CD). In this work, we designed a small library of AIEC antiadhesives. A lead compound was shown to decolonize AIEC from the gut of a transgenic mouse model of CD after oral administration. The water-soluble and non-cytotoxic compounds developed may open new perspectives in the treatment of CD.
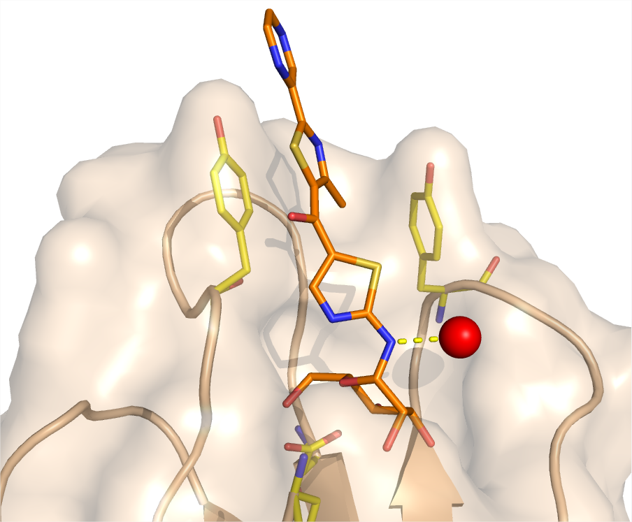
 4 - Thiazolylaminomannosides as potent anti-adhesives of type 1 piliated Escherichia coli isolated from Crohn’s disease patients.
Brument, S.; Sivignon, A.; Dumych, T. I.; Moreau, N.; Roos, G.; Guérardel, Y.; Chalopin, T.; Deniaud, D.; Bilyy,R. O. ; Darfeuille-Michaud, A.; * Bouckaert, J.; Gouin, S. G. J. Med. Chem, 2013, 56, 5395-5406
Adherent-invasive Escherichia coli (AIEC) have previously been shown to induce gut inflammation in patients with Crohn’s disease (CD). We developed a set of mannosides to prevent AIEC attachment to the gut by blocking the FimH bacterial adhesin. The crystal structure of the FimH lectin domain in complex with a lead thiazolylaminomannoside highlighted the preferential position for pharmacomodulations. A small library of analogues showing nanomolar affinity for FimH was then developed.
Notably, AIEC attachment to intestinal cells was efficiently prevented by the most active compound and at around 10000-fold and 100-fold lower concentrations than mannose and the potent FimH inhibitor heptylmannoside, respectively.
An ex vivo assay performed on the colonic tissue of a transgenic mouse model of CD confirmed this antiadhesive potential. Given the key role of AIEC in the chronic intestinal inflammation of CD patients, these results suggest a potential antiadhesive treatment with the FimH inhibitors developed.
4 - Thiazolylaminomannosides as potent anti-adhesives of type 1 piliated Escherichia coli isolated from Crohn’s disease patients.
Brument, S.; Sivignon, A.; Dumych, T. I.; Moreau, N.; Roos, G.; Guérardel, Y.; Chalopin, T.; Deniaud, D.; Bilyy,R. O. ; Darfeuille-Michaud, A.; * Bouckaert, J.; Gouin, S. G. J. Med. Chem, 2013, 56, 5395-5406
Adherent-invasive Escherichia coli (AIEC) have previously been shown to induce gut inflammation in patients with Crohn’s disease (CD). We developed a set of mannosides to prevent AIEC attachment to the gut by blocking the FimH bacterial adhesin. The crystal structure of the FimH lectin domain in complex with a lead thiazolylaminomannoside highlighted the preferential position for pharmacomodulations. A small library of analogues showing nanomolar affinity for FimH was then developed.
Notably, AIEC attachment to intestinal cells was efficiently prevented by the most active compound and at around 10000-fold and 100-fold lower concentrations than mannose and the potent FimH inhibitor heptylmannoside, respectively.
An ex vivo assay performed on the colonic tissue of a transgenic mouse model of CD confirmed this antiadhesive potential. Given the key role of AIEC in the chronic intestinal inflammation of CD patients, these results suggest a potential antiadhesive treatment with the FimH inhibitors developed.
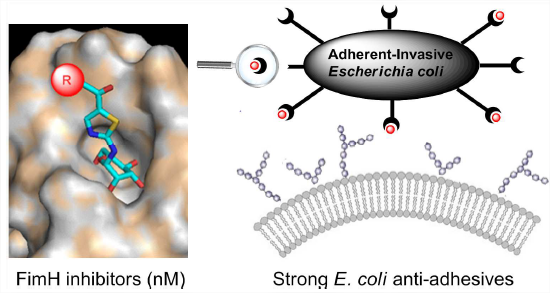 5 - Heptyl α-D-mannosides grafted on a β-cyclodextrin core to interfere with Escherichia coli adhesion – an in vivo multivalent effect.
Bouckaert, J.; Zhaoli; Li.; Xavier, C.; Almant, M.; Caveliers; V.; Lahoutte, T.; Weeks, S.; Kovensky, J; Gouin, S.G. Chem. Eur. J. 2013, 24, 7847-7855.
Let’s stick together. One pot “click or co-clicking” procedures were developed to directly obtain mono- and heptavalent conjugates from unprotected alkynyl-armed heptyl mannoside (HM) and azido--CD synthons.
These anti-adhesive were shown to aggregate both the FimH adhesin and uropathogenic bacteria in solution. The first in vivo evaluation of synthetic multivalent FimH inhibitors was performed. The heptavalent -CD proved to be much more effective anti-adhesive agents than monovalent HM references, with doses of around 2 μg instilled in the mouse bladder leading to a significantly decreased E. coli load. Intravenously injected radiolabelled glycoconjugate can rapidly reach the mouse bladder and are retained over 24 h.
5 - Heptyl α-D-mannosides grafted on a β-cyclodextrin core to interfere with Escherichia coli adhesion – an in vivo multivalent effect.
Bouckaert, J.; Zhaoli; Li.; Xavier, C.; Almant, M.; Caveliers; V.; Lahoutte, T.; Weeks, S.; Kovensky, J; Gouin, S.G. Chem. Eur. J. 2013, 24, 7847-7855.
Let’s stick together. One pot “click or co-clicking” procedures were developed to directly obtain mono- and heptavalent conjugates from unprotected alkynyl-armed heptyl mannoside (HM) and azido--CD synthons.
These anti-adhesive were shown to aggregate both the FimH adhesin and uropathogenic bacteria in solution. The first in vivo evaluation of synthetic multivalent FimH inhibitors was performed. The heptavalent -CD proved to be much more effective anti-adhesive agents than monovalent HM references, with doses of around 2 μg instilled in the mouse bladder leading to a significantly decreased E. coli load. Intravenously injected radiolabelled glycoconjugate can rapidly reach the mouse bladder and are retained over 24 h.

Multivalent ligands are extensively used to inhibit carbohydrate-binding proteins (lectins). We have transferred this concept to carbohydrate-processing enzymes (glycosidases), by synthesizing and showing that multivalent transition-state inhibitord could inhibit or boost enzymatic activities.
 Selected articles
Selected articles
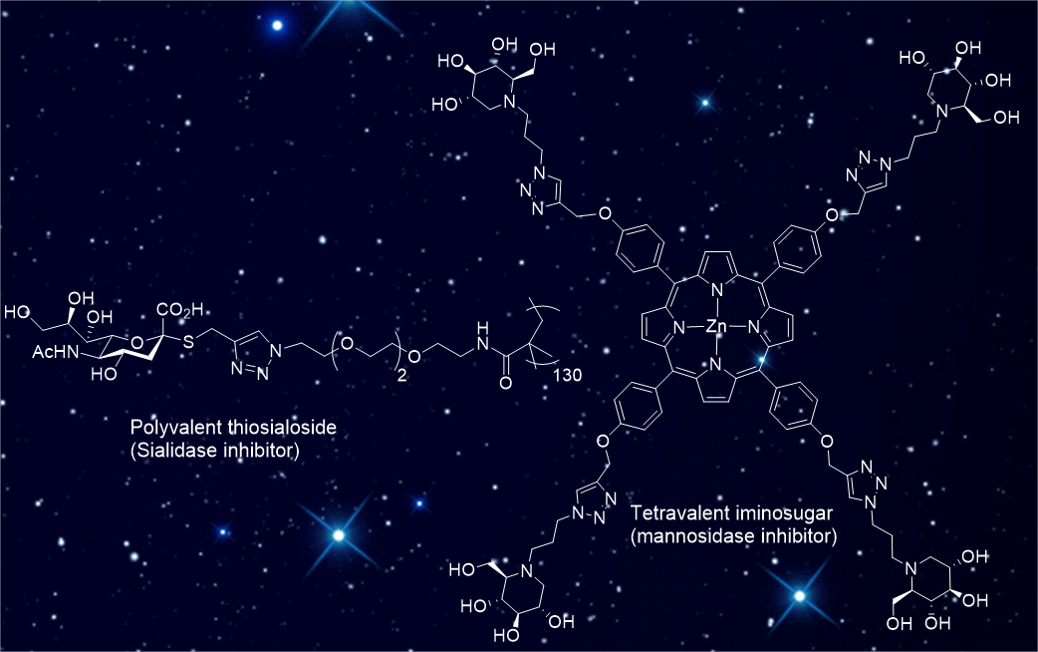
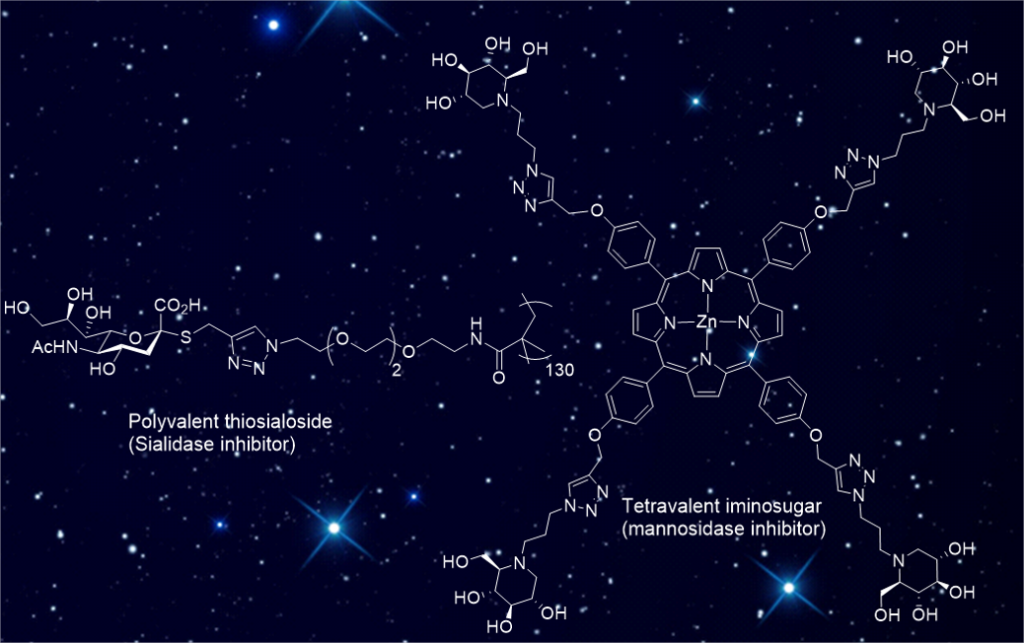
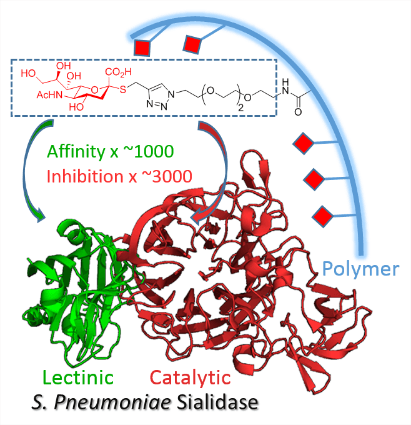
1 - Multivalent Thiosialosides and Their Synergistic Interaction with Pathogenic Sialidases.
Brissonnet, Y. ; Assailly, C.; Saumonneau, A. ; Bouckaert, J.; Maillasson, M.; Petitot, C.; Roubinet, B.; Didak, B.; Landemarre, L.; Bridot, C.; Blossey, R.; Deniaud, D.; Yan, X.; Bernard, J.; Tellier, C.; Grandjean, C.; Daligault, F.; Gouin S.G. Hot Paper, Chem. Eur. J. 2019, 25, 2358-2365
High synergy: Sialidases (SA) are virulence factors expressed by pathogenic bacteria, viruses and parasites. Di- and polymeric thiosialosides were designed to inhibit V. cholerae, T. cruzi and S. pneumoniae sialidases. Each thiosialoside grafted on the polymer was shown to reduce SA catalytic activity with much higher efficiency compared to their monovalent analogues. This extend the multivalent concept to this class of enzymes.
2 - Magnetic nanoparticules coated with thiomannosides or iminosugars to switch and recycle galactosidase activity;
Alvarez-Dorta, D.; Brissonnet, Y.; Saumonneau, A.; Deniaud, D.; Bernard, J.; Tellier, C. ; Daligault, F. ;Gouin, S.G. * Chemistry Select, 2017, 2, 9552-9556.
Speed 2. Magnetic nanoparticles coated with thiomannosides (SMan@Fe3O4) and deoxynojirimycin iminosugars (DNJ@Fe3O4) switched α-galactosidase (αGal) activity from strong inhibition to activation profiles, respectively. The αGal-DNJ@Fe3O4 complex could be magnetically recovered and conserve a higher catalytic velocity than αGal alone during the first four cycles.

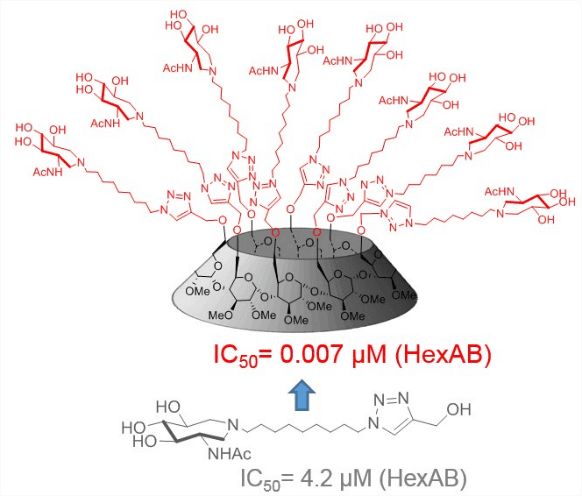
3 - Multivalency to potently inhibit and discriminate between hexosaminidase.
Alvarez-Dorta, D.; King, D.; Legigan, T.; Kato, A.; Désiré, J.; Deniaud, D.; Vocadlo, D.; Gouin,* S.G.; Blériot,* Y. Chem. Eur.J. 2017, 23, 9022-9025.
All crowded round: Multivalent acetamidoazepanes were designed to assess the effect of multivalency against biologically relevant β-hexosaminidases. Nanomolar multivalent inhibitors of HexAB, with improved enzyme selectivity, could be obtained by clustering a micromolar azepan inhibitor. These results extend the multivalent concept to an important class of glycosidases.
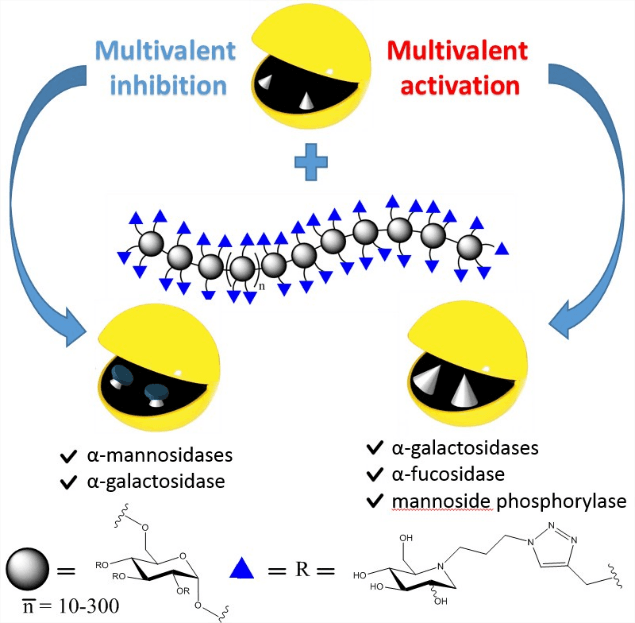
4 - Polymeric iminosugars to improve the activity of carbohydrate-processing enzymes.
Brissonnet,Y.; Tezé, D.; Fabre, E.; Deniaud, D.; Daligault, F.; Tellier, C.; Šesták, S.; Ladévèze, S.; Remaud-Simeon, M.; Potocki- Véronèse, G.; Gouin, S. G. * Bioconjugate chem. 2015, 26, 766-772.
Multivalent iminosugars have recently emerged as powerful tools to inhibit the activities of specific glycosidases. In this work, biocompatible dextrans were coated with iminosugars to form linear and ramified polymers with unprecedently high valencies (from 20 to 900) to probe the evolution of the multivalent inhibition as a function of ligand valency.
This study led to the discovery that polyvalent iminosugars can also significantly enhance, and not only inhibit, the enzymatic activity of specific glycoside-hydrolase, as observed on two galactosidases, a fucosidase, and a bacterial mannoside phosphorylase for which an impressive 70-fold activation was even reached. The concept of glycosidase activation is largely unexplored, with a unique recent example of small-molecules activators of a bacterial O-GlcNAc hydrolase. The possibility of using these polymers as “artificial enzyme effectors” may therefore open up new perspectives in therapeutics and biocatalysis.
5 - Topological effects and binding modes operating with multivalent iminosugar-based glycoclusters and glycosidases.
Brissonnet Y., Ortiz-Mellet C., Morandat S., Garcia Moreno I., Deniaud D., Matthews S. E., Vidal S., El Kirat K., Gouin. S. G. * J. Am. Chem. Soc. 2013, 135, 18427-18435.
Multivalent iminosugars have been recently explored for glycosidase inhibition. Affinity enhancements due to multivalency have been reported for specific targets, which are particularly appealing when a gain in enzyme selectivity is achieved but raise the question of the binding mode operating with this new class of inhibitors.
Here we describe the development of a set of tetra- and octavalent iminosugar probes with specific topologies and an assessment of their binding affinities toward a panel of glycosidases including the Jack Bean α-mannosidase (JBαMan) and the biologically relevant class II α-mannosidases from Drosophila melanogaster belonging to glycohydrolase family 38, namely Golgi α-mannosidase ManIIb (GM) and lysosomal α-mannosidase LManII (LM). Very different inhibitory profiles were observed for compounds with identical valencies, indicating that the spatial distribution of the iminosugars is critical to fine-tune the enzymatic inhibitory activity.

Compared to the monovalent reference, the best multivalent compound showed a dramatic 800-fold improvement in the inhibitory potency for JBαMan, which is outstanding for just a tetravalent ligand. The compound was also shown to increase both the inhibitory activity and the selectivity for GM over LM.
This suggests that multivalency could be an alternative strategy in developing therapeutic GM inhibitors not affecting the lysosomal mannosidases. Dynamic light scattering experiments and atomic force microscopy performed with coincubated solutions of the compounds with JBαMan shed light on the multivalent binding mode. The multivalent compounds were shown to promote the formation of JBαMan aggregates with different sizes and shapes. The dimeric nature of the JBαMan allows such intermolecular cross-linking mechanisms to occur.
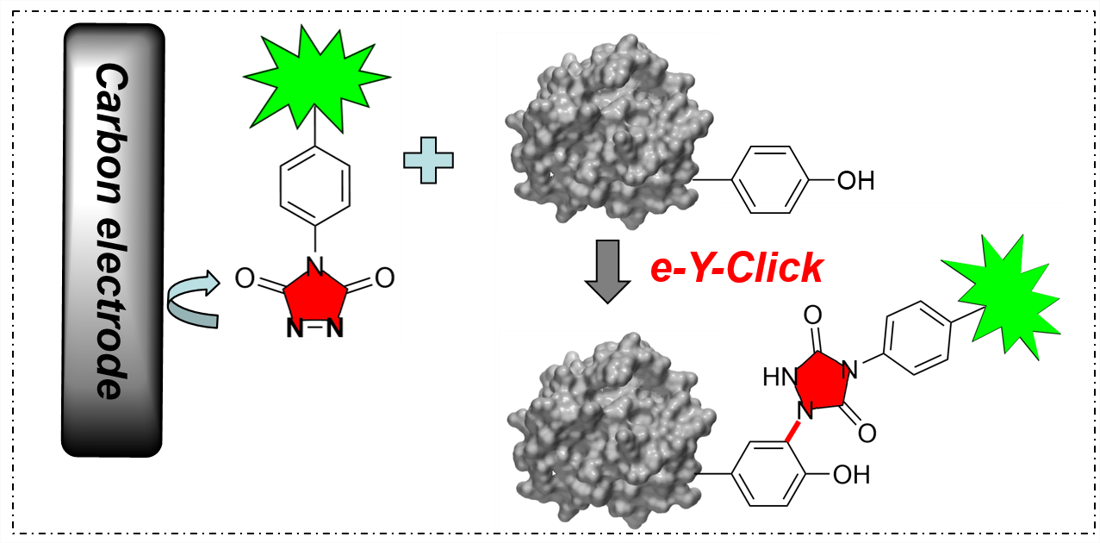
We recently developed the first electrochemical bioconjugation method for protein labelling (see figure). We are also interested in improving radiolabeling strategies for cancer imaging, therapy and metal decorporation.
Five selected articles
Electrochemically promoted tyrosine-click-chemistry for protein labelling
Dimitri Alvarez-Dorta, Christine Thobie-Gautier, Mikael Croyal, Mohammed Bouzelha, Mathieu Mével, David Deniaud, Mohammed Boujtita, and Sébastien G. Gouin. J. Am. Chem. Soc. 2018, 140, 17120.
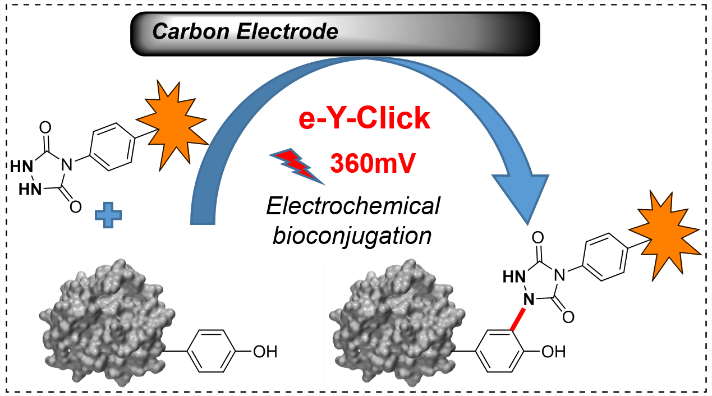 The development of new bioorthogonal ligation methods for the conjugation of native proteins is of particular importance in the field of chemical biology and biotherapies. In this work, we developed a traceless electrochemical method for protein bioconjugation. The electrochemically promoted tyrosine-click (e-Y-CLICK) allowed the chemoselective Y-modification of peptides and proteins with labeled urazols. A low potential is applied in an electrochemical cell to activate PTAD anchors in situ and on demand, without affecting the electroactive amino-acids from the protein. The versatility of the electrosynthetic approach was shown on biologically relevant peptides and proteins such as oxytocin, angiotensin 2, serum bovine albumin and epratuzumab. The fully conserved enzymatic activity of a glucose oxidase observed after e-Y-CLICK further highlights the softness of the method. The e-Y-CLICK protocols were successfully performed in pure aqueous buffers, without the need for co-solvents, scavenger or oxidizing chemicals, and should therefore significantly broaden the scope of bioconjugation.
The development of new bioorthogonal ligation methods for the conjugation of native proteins is of particular importance in the field of chemical biology and biotherapies. In this work, we developed a traceless electrochemical method for protein bioconjugation. The electrochemically promoted tyrosine-click (e-Y-CLICK) allowed the chemoselective Y-modification of peptides and proteins with labeled urazols. A low potential is applied in an electrochemical cell to activate PTAD anchors in situ and on demand, without affecting the electroactive amino-acids from the protein. The versatility of the electrosynthetic approach was shown on biologically relevant peptides and proteins such as oxytocin, angiotensin 2, serum bovine albumin and epratuzumab. The fully conserved enzymatic activity of a glucose oxidase observed after e-Y-CLICK further highlights the softness of the method. The e-Y-CLICK protocols were successfully performed in pure aqueous buffers, without the need for co-solvents, scavenger or oxidizing chemicals, and should therefore significantly broaden the scope of bioconjugation.
Selected articles
Investigation of a new "N2S2O2" chelating agent with high Po(IV) affinity
Younes A.; Montavon G.; Gouin S.G.; André-Joyaux E.; Peumery R.; Chalopin T.; Alliot C.; Mokili M.; Champion J.; Deniaud D. Chem. Comm., 2017, 53, 6492-6495.
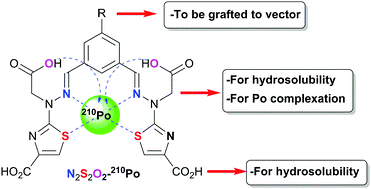
A water-soluble “N2S2O2” complexing agent was designed for polonium(IV) decorporation. The bifunctional ligand showed outstanding Po(IV) complexing abilities, with a conditional stability constant three orders of magnitude higher than the reference ligand BAL.
Targeted radionuclide therapy with astatine-211: Oxidative dehalogenation of astatobenzoate conjugates
Teze D.; SergentuD-C.; Kalichuk V.; Barbet J.; Deniaud D.; Galland N.; Maurice R.; Montavon G. Scientific Reports, 2017, 7, article number 2579.

At is a most promising radionuclide for targeted alpha therapy. However, its limited availability and poorly known basic chemistry hamper its use. Based on the analogy with iodine, labelling is performed via astatobenzoate conjugates, but in vivo deastatination occurs, particularly when the conjugates are internalized in cells. Actually, the chemical or biological mechanism responsible for deastatination is unknown. In this work, we show that the C−At "organometalloid" bond can be cleaved by oxidative dehalogenation induced by oxidants such as permanganates, peroxides or hydroxyl radicals. Quantum mechanical calculations demonstrate that astatobenzoates are more sensitive to oxidation than iodobenzoates, and the oxidative deastatination rate is estimated to be about 6 × 106 faster at 37 °C than the oxidative deiodination one. Therefore, we attribute the “internal” deastatination mechanism to oxidative dehalogenation in biological compartments, in particular lysosomes.
Recent advances in cancer treatment by iron chelators (review)
Corcé V.; Gouin S.G.; Renaud S.; Gaboriau F.; Deniaud D. Bioorg. Med. Chem. Lett. 2016, 26, 251-256.
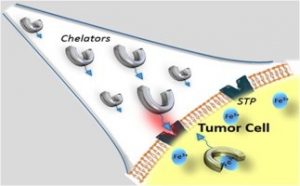
The development of new therapeutic alternatives for cancers is a major public health priority. Among the more promising approaches, the iron depletion strategy based on metal chelation in the tumoral environment has been particularly studied in recent decades. After a short description of the importance of iron for cancer cell proliferation, we will review the different iron chelators developed as potential chemotherapeutics. Finally, the recent efforts to vectorize the chelating agents specifically in the microtumoral environment will be discussed in deta.
Synthesis and characterization of a stable copper(I) complex for radiopharmaceutical applications
Bodio E.; Boujtita M.; Julienne K.; Le Saec P.; Gouin S.G.; Hamon J.; Renault E.; Deniaud D. ChemPlusChem, 2014, 79, 1284-1293.
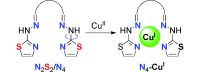
N4Cu complexes: A series of ligands has been synthesized and their complexation with copper(II) precursors studied. The combination of several analytical techniques and density functional calculations shows that N4 copper(I) complexes are formed from a copper(II) salt (see figure). Preliminary tests of complexation show the ability of these ligands to chelate 64Cu in very diluted medium, which is of interest for developing new positron emission tomography imaging agents.
Our group also focuses on the development of novel, chemically modified adeno-associated virus (AAV) particles, with increased therapeutic efficiency, at the interface between vectorology and chemistry. AAVs are therapeutic platforms for the treatment of genetic diseases. Yet, past and ongoing clinical trials have highlighted some limitations, including the need for high doses to achieve therapeutic benefit, and the off-target transduction of various tissues. Chemically modified AAVs (ϕ-AAV) greatly decrease off-target effects, hence maximizing transduction in tissues of interest and improving the therapeutic index in AAV-based gene therapy. This was achieved by the covalent coupling of tissue-specific ligands on surface exposed residues of the AAV capsid. The main advantage of this chemistry oriented approach is to be able to functionalize AAV particles with various ligands which could not be genetically encoded, including polymers, sugars or lipids.

Project 1: Chemical modification of amino group on the surface of the capsid of AAV for hepatocytes targeting
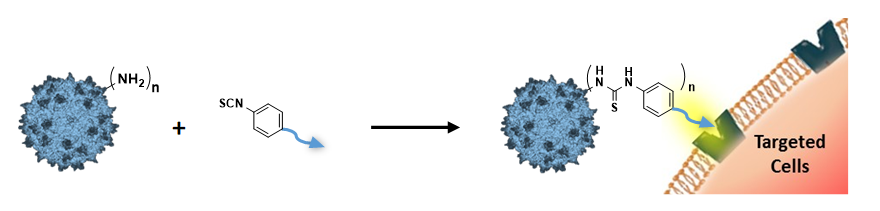
These modifications are achieved by chemical coupling of a ligand by the formation of a thiourea functionality between the amino group of the capsid proteins and the reactive isothiocyanate motif incorporated into the ligand. This strategy does not require genetic engineering of the capsid sequence. The proof of concept was first evidenced using a fluorophore (FITC). Next, we coupled the N-acetylgalactosamine ligand onto the surface of the AAV capsid for asialoglycoprotein receptor-mediated hepatocyte-targeted delivery. Chemically-modified capsids showed reduced interactions with neutralizing antibodies. Taken together, our findings reveal the possibility of creating a specific engineered platform for targeting AAVs via chemical coupling.
Project 2: Chemical modification of tyrosine group on the surface of the capsid of AAV
This project investigate the possibility of re-targeting rAAV vectors via covalent linking of specific ligands to the tyrosine of AAV capsid by taking advantage of click-tyrosine chemical modification. Indeed it has been demonstrated that surface exposed tyrosine mutation on AAV2 capsids avoided degradation by the proteasome and resulted in high-efficiency transduction in human cells in vitro and murine hepatocytes in vivo. This “enhancer effect”, obtained after tyrosine mutation, has only been observed for this amino-acid and not after lysine mutation. These results demonstrated the prominent role of rAAV capsid tyrosine in cellular trafficking and transgene expression. This part will focus on the bioconjugaison of click tyrosine ligand, having specific targeting properties, on the capsid of AAV in one step. The proof of concept data has been protected by a patent filled in 2019.

Selected article
Chemical modification of the adeno-associated virus capsid to improve gene delivery Mathieu Mével,* Mohammed Bouzelha, Aurélien Leray, Simon Pacouret, Mickael Guilbaud, Magalie Penaud-Budloo, Dimitri Alvarez-Dorta, Laurence Dubreil, Sébastien G. Gouin, Jean Philippe Combal, Mirja Hommel, Gloria Gonzalez-Aseguinolaza, Véronique Blouin, Philippe Moullier, Oumeya Adjali, David Deniaud,* Eduard Ayuso.* Chem. Sci. 2019, 140, 17120.
Gene delivery vectors based on adeno-associated virus (AAV) are highly promising due to several desirable features of this parent virus, including a lack of pathogenicity, efficient infection of dividing and non-dividing cells and sustained maintenance of the viral genome. However, the conclusion from clinical data using these vectors is that there is a need to develop new AAVs with a higher transduction efficiency and specificity for relevant target tissues. To overcome these limitations, we chemically modified the surface of the capsid of AAV vectors. These modifications were achieved by chemical coupling of a ligand by the formation of a thiourea functionality between the amino group of the capsid proteins and the reactive isothiocyanate motif incorporated into the ligand. This strategy does not require genetic engineering of the capsid sequence. The proof of concept was first evidenced using a fluorophore (FITC). Next, we coupled the N-acetylgalactosamine ligand onto the surface of the AAV capsid for asialoglycoprotein receptor-mediated hepatocyte-targeted delivery. Chemically-modified capsids also showed reduced interactions with neutralizing antibodies. Taken together, our findings reveal the possibility of creating a specific engineered platform for targeting AAVs via chemical coupling. 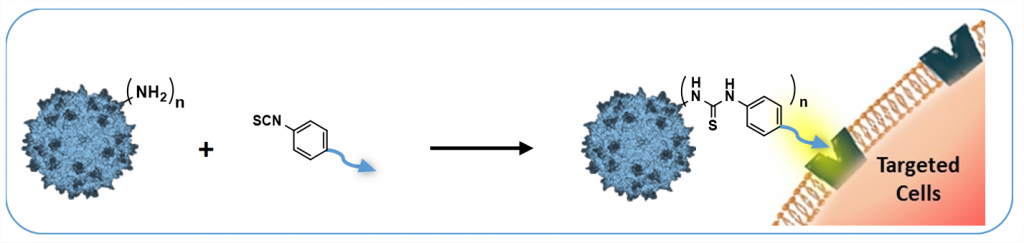
Team and academic partners







| Dr Mohammed Boujtita, Laboratory CEISAM, Univ. Nantes, (Electrochemistry) | Dr Ayuso Eduard, INSERM UMR 1089, Nantes, (Virus modification) | Dr Julie Bouckaert, UGSF UMR 8576, Univ. Lille, (Biophysical analysis) |
| Dr Patrice Le Pape, IICiMed, CHU Nantes, (A. fumigatus assays) | Dr Annabelle Varrot, CERMAV, Grenoble (ITC, SPR) | Dr Nicolas Barnich, INSERM UMR 1071, Clermont-Ferrand (Crohn’s disease models) |
| Dr Ludovic Landemarre, GLYCODIAG, Orléans (Binding assays) | Dr Franck Daligault, UFIP, UMR 6286, Nantes (Enzymatic assays) | Dr Eva-Maria Krammer, UGSF UMR 8576, Univ. Lille, (Docking, molecular simulations) |
| Prof Yves Bleriot, IC2MP, UMR 7285, Poitiers (Iminosugars) | Prof Sébastien Papot, IC2MP, UMR 7285, Poitiers (Iminosugars) | Prof Beat Ernst, Pharmacenter, CH (lectin antagonists) |
| Dr Rostyslav Bilyy, Medical Univ., Lviv, Ukraine (Biological assays) | Dr Benjamin Bouvier, LG2A, UMR 7378., Amiens (In silico molecular modeling) | Dr Gilles Montavon, Subatech, IMT Atlantique, Nantes (Radiochemistry) |
| Dr Nicolas Galland, CEISAM, UMR 6230, Nantes, (Modelisation) | Dr Caroline Le Guiner, INSERM UMR 1089, Nantes (Gene therapy) | Dr Jérôme Guicheux, RMeS, UMR 1229, Nantes, (Regenerative Medecine) |
| Dr Mikaël Croyal, INRAE, UMR 1280, Nantes, (Mass spectroscopy) | Dr. Els Verhoeyen, ENS Lyon, UMR 1111, CIRI, Lyon, (Cell Biology) | Prof. Ferid Haddad, GIP Arronax, Nantes, (Radiochemistry) |
Financials
![]()

![]()

![]()