Cette thématique regroupe tous les développements méthodologiques de l’équipe ModES qu’il s’agisse de logiciels ou de l’élaboration de protocoles afin de traiter des systèmes particuliers.
The bonding in many compounds containing heavy atoms is still unclear, especially because of relativistic effects and in particular the spin-orbit coupling (SOC). The latter complicates the possibility of imagining a bridge between the complex electronic structure and simple concepts, such as covalent and ionic bonds, single and multiple bonds, VSEPR structure, etc. In this perspective, we are developing various innovative tools to probe the effects of SOC on the chemical bond.
In this perspective, we develop various innovative tools for probing the SOC effects on the chemical bonding. The topological analysis of the electron localization function (ELF) and the Bader’s theory of atoms in molecules (QTAIM), were introduced into the formalism of two-component relativistic DFT calculations (Figure below). Regarding the molecular orbital theory, the effective bond order (EBO) concept was extended to multi-configurational wave functions at the spin-orbit configuration interaction level (SOCI).


The team is also developing functional benchmarks tests as part of the simulation of optical spectra. More specifically, we were interested, in collaboration with the teams of Carlo Adamo (Paris) and Benedetta Mennucci (Pisa) in the energies of 0-0 transitions as well as in the shapes of the absorption and emission bands. The objective was to answer several questions: a) what is the most suitable functional for a particular spectroscopic characteristic ? b) what precision can be expected? c) is this precision uniform for all the families of transitions present in organic compounds?
We also study the strengths and weaknesses of the GW / BSE approach from solid state physics as part of the modeling of excited states of molecules. This work, carried out in collaboration with the team of Xavier Blase (Grenoble) made it possible, among other things, to show the interest of this approach for the states presenting a strong multiplied excited character.
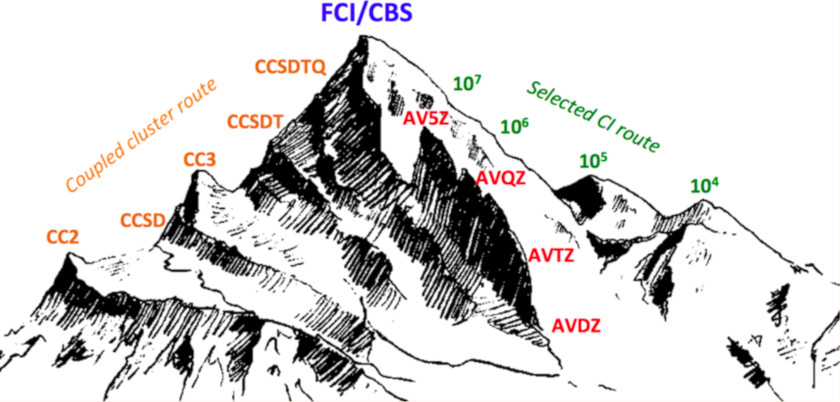
Finally, in collaboration with Dr. Pierre-François Loos, we define very precise vertical transition energies by combining Coupled-Cluster and selected CI approaches. The goal is to have an undeniable set of quality values at hand so that we can objectively assess lighter methods. These approaches are also used to achieve chemical precision for energies 0-0.
Reference publications:
TD-DFT related:
- D. Laurent, D. Jacquemin Int. J. Quantum Chem. 2013, 113, 2019-39.
- A. Charaf-Eddin et al. J. Chem. Theory Comput.2013, 9, 2749-2760.
BSE related:
- Jacquemin, I. Duchemin, X. Blase J. Chem. Theory Comput. 2015, 11, 5340-5359.
- D. Jacquemin, I. Duchemin, X. Blase J. Chem. Theory Comput. 2015, 11, 3290-3304.
- D. Jacquemin, I. Duchemin, X. Blase Chem. Soc. Rev. 2018, 47, 1022-1043.
High-accuracy related :
COPReMo (COlor PRediction of Molecules): python package for predicting color from an absorption or emission spectrum.
CT (Charge Transfer): Fortran code to obtain the electronic density difference between states and to calculate charge transfer parameters from cube files generated by Gaussian.