Personnes impliquées : Anais Goupille (AI), Jérôme Graton (MC), Adèle Laurent (CR) Jean-Yves Le Questel (PR), Néha Tripathi (Post-doctorante).
Présentation générale
Dans le cadre de cette thème, nos travaux sont essentiellement consacrés à l’étude d’interactions moléculaires de type non-covalentes, en particulier la liaison hydrogène (liaison H) par des approches combinant des observations expérimentales et des études théoriques sur des systèmes croissants en taille et en complexité. Des études quantitatives sont ainsi effectuées sur des molécules organiques simples (axe 1), permettant de sonder de façon très fine différents effets, en particulier l’influence de l’environnement intramoléculaire d’un site accepteur/donneur de liaison H sur les caractéristiques des interactions établies.
Un second thème, fondé sur la mise en œuvre des mêmes approches, est consacré à la détermination de l’influence de la fluoration de composés organiques sur leurs propriétés (potentiel d’interaction, lipophilie). Un troisième axe se focalise sur l’élucidation des interactions de modulateurs compétitifs des récepteurs nicotiniques d’insectes (insecticides néonicotinoïdes et leurs derniers dérivés) avec leur cible. Le dernier thème, dédié aux interactions protéine-protéine, fondé sur de nombreuses collaborations, a en particulier pour objectif de concevoir de façon rationnelle des modulateurs (activateurs, inhibiteurs) des cibles visées.
Despite the very large number of works devoted to H-bond interactions in the literature, experimental quantitative energy data, reflecting the structural and chemical diversity encountered in organic molecules, are still lacking. From experimental and theoretical studies of a range of compounds containing functional groups relevant in organic (medicinal) chemistry, we measure a set of thermodynamic and spectroscopic parameters related to their H-bond strength.
The differences observed in terms of behavior are in parallel investigated by quantum chemistry calculations. In recent years, our work within the framework of this thematic has in particular been devoted to new chemical patterns belonging to heterocycles (see figure opposite) and to the quantification of the H-bonding acidity (pKAHY) of selected samples of organic compounds.
The objective of this axis is to make available to the scientific community a set of parameters describing in a precise and homogeneous manner the H-bond basicity and acidity of functional groups relevant in organic (medicinal) chemistry. The success of the pKBHX database, developed ten years ago, both at the industrial and academic levels, has clearly confirmed the lack of this type of data and their interest for a large community, in particular in medicinal chemistry, but also in organic and supramolecular chemistry.
The fluorination of organic compounds is of interest for the optimization of diverse properties (chemical, physical, pharmacological) of molecules and finds applications in most fields of the chemical industry. Paradoxically, studies aimed at rationalizing the influence of fluorine introduction in organic compounds are relatively few. Our work within the framework of this project is at the heart of this context. As part of a collaboration established for several years with the group of Prof. Bruno Linclau, at the University of Southampton, specialized in the synthesis of fluorinated organic molecules, we study, through an approach combining experimental measurements and theoretical calculations, the influence of the introduction of fluorine atom (s) on the physicochemical properties (potential of interactions, lipophilicity ...) of organic compounds.
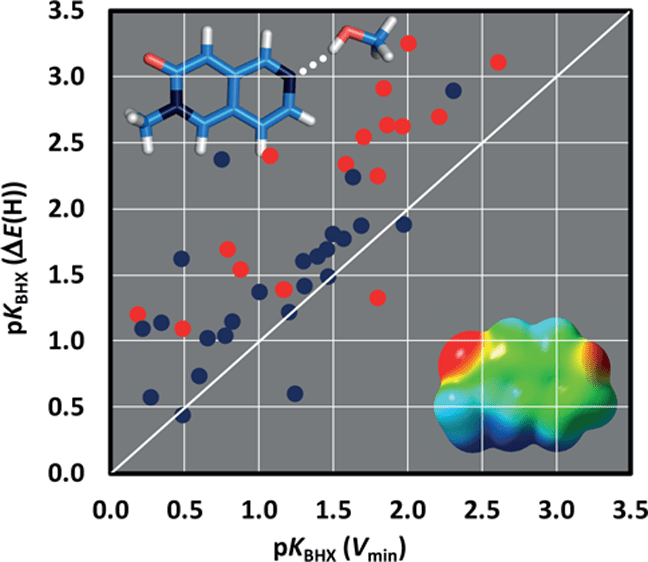
As an example, we have recently shown (see figure opposite), thanks to the study of flexible molecules, that the sometimes unexpected differences in terms of pKAHY between close diastereoisomers can be rationalized on the basis of subtle populations variations of conformers in which interactions by intramolecular OH ••• F H-bonds occur, as well as by the strength of these interactions. From these results, a relationship between pKAHY and a variant of the molecular electrostatic potential, extended to fluorohydrins, has been proposed, this calibration line now being established on a sample of nearly 80 hydroxylated compounds. This study clearly underlines the importance of weak OH ••• F intramolecular H-bond interactions in the modulation of an OH group H-bond acidity.

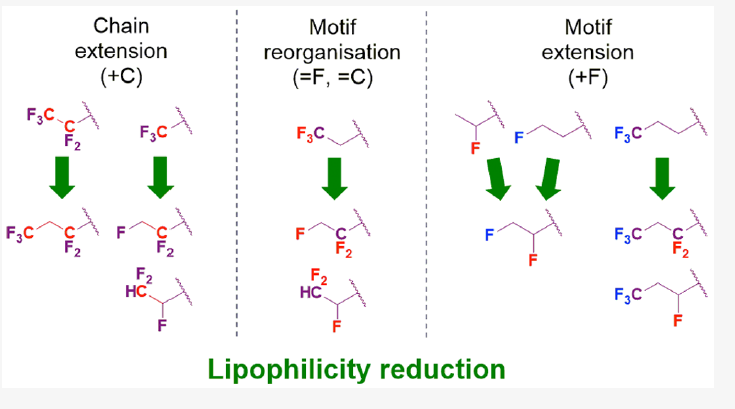
This work also makes it possible to characterize the lipophilicity potential of fluorinated organic compounds. Thus, through a systematic investigation of the modulation of lipophilicity following the introduction of different fluorination patterns, we were able to precisely quantify the effect of specific patterns. This work therefore offers the possibility of proposing avenues to rationally modulate the properties of organic molecules through the introduction of specific fluorination patterns (number and relative position of fluorine atoms).
This project presents applications in a large number of fields in chemistry. Our objective is, ultimately, to develop a set of tools and rules allowing a quantitative prediction and a detailed description of the effect of the introduction of fluorine atom(s) on fundamental physicochemical properties of molecules containing functionalities encountered in organic ligands with therapeutic aims.
Despite the restrictions on the use of neonicotinoids in Europe due to their role in the decline of pollinating insects, their persistence in the environment continues to raise many questions in terms of toxicities, including potentially for mammals and humans, due to their accumulation and the possibility of cocktail effects.
In collaboration with Pr. Steeve Thany, neurobiologist and electrophysiologist at the University of Orléans, we elucidate the interactions of neonicotinoids and their recent derivatives (in particular sulfoxaflor, flupyradifurone) with a model of the extracellular ligand binding domain of insects nicotinic acetylcholine receptors (nAChRs).
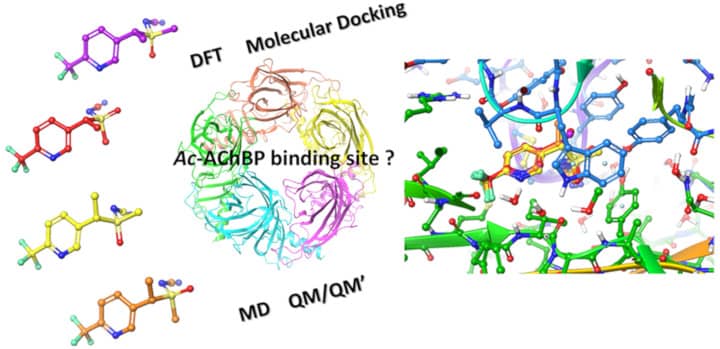
For sulfoxaflor (figure opposite), this work has thus shown the importance of the stereochemistry of the compound in the interaction with the active site of the receptor, very different networks of interactions being brought into play according to the ligand diastereoisomers. The advantage of using different methods: quantum mechanics (DFT), molecular docking, Molecular Dynamics and finally hybrid QM/QM' methodologies was also highlighted, so as to pinpoint with precision, both on a structural and energetic level, the interactions involved.
Ultimately, this work, carried out at different scales on nAChRs of bees and mammals, should lead to a more precise understanding of the toxic effect of these compounds.
The prediction of protein-protein interactions and their use to develop new drugs is currently an important area of research in medicinal chemistry. In the context of work carried out in collaboration with different teams of biologists, organic chemists and modellers, our work aims to describe and rationalize the interactions between inhibiting compounds of particular biological receptors in order to i) determine their binding sites, ii) explain experimental binding affinity data and / or iii) identify new inhibitors targeting a specific binding site of a macromolecule. To this end, virtual screening and classical molecular dynamics (MD) techniques have been used systematically.
About the study of the bioavailability of a series of Rad51 polymerase inhibitors, we have thus shown that, depending on the nature of the functional groups in para position of disulfonic stilbene derivatives, their preferential binding site to human serum albumin (key protein to consider in ADME processes) is very different. Indeed, the interaction between each of the derivatives and each of the four binding sites has been characterized in structural terms, specific interactions or even energy affinity (MM-GB/PBSA). We have shown, in agreement with experience, that the flagship inhibitor with two isothiocynanate functions has a high / low affinity for the IIIA / IB binding site, confirming the presence of a second binding site observed experimentally (isothermal titration calorimetry (ITC), circular dichroism and fluorescence).
In a methodological context, we also compared several molecular screening approaches to classical MD to understand the IC50 values from the literature of cyclic inhibitors of the CD2-CD58 interaction, playing a role in the prevention of the immune response. Due to the great flexibility of known inhibitors, the simulation of interactions with the differentiation cluster (CD58) in virtual screening has shown its confines, while conventional simulations have made it possible to finely understand and rationalize the different affinities with CD58. [J. Comput. Aid. Mol. Des. (2019), 32 1295.]
Finally, as part of an interdisciplinary project, we, in partnership with CRCINA biologists, undertook an in-depth study of the structural characteristics of interleukin-15 (IL-15). IL-15 plays an important role in the communication between cells of the immune system and has emerged as a potential therapeutic target for different therapeutic applications. The structure of this cytokine is in fact based on a quaternary complex between IL-15 and its alpha (IL-15Ra), beta (IL-2Rb) and gamma (gc) receptors. The functional modulation of IL-15 is linked to its interaction with these receptors, in particular IL-2Rb. We will illustrate here the work carried out in this context by the results provided by the implementation of a joint approach coupling DM simulations, studies of directed mutagenesis and functional measurements. We have thus updated the significant contribution of a new amino acid of IL-15 (Ser58, S58, see Figure below)) in the interaction with the IL-2Rb receptor.
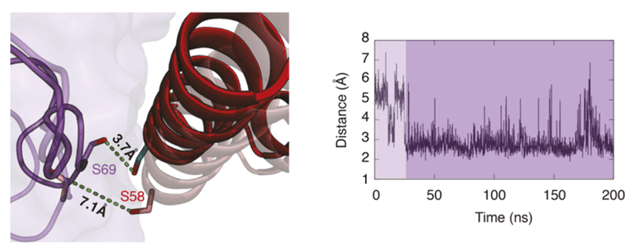
This highlighting is specially remarkable since previous studies by X-ray crystallography had not revealed any particular behavior for this amino acid. Thus, the highly labile and dynamic nature of the protein-protein interfaces suggests supplementing the experimental and / or theoretical studies with MD studies. Our simulations thus show that on the first 30 ns of simulations, no interaction is detectable between the residues Ser58 located on helix C of IL-15 (below in red) and Ser69 of IL- 2Rb (in purple). A conformational transition occurs however around 40 ns, an hydrogen-bond interaction being established between the two residues, the interaction persisting practically throughout the duration of the simulation.
The study of other mutants, notably combined with surface plasmon resonance (SPR) measurements and studies of signal transduction (expression of p-STAT5) confirmed the loss of signaling for some of them.
Collaborations de recherche
- Sophie Brouard (Université de Nantes) Centre de Recherche en Transplantation et Immunologie de Nantes, Équipe 4 : Immunoregulation and Immunointerventions in Trans-plantation and Autoimmunity. (lien)
- Fabrice Fleury (Université de Nantes) Unité Fonctionnalité et ingénierie des Protéines (UFIP) UMR 6286, équipe Mécanisme et régulation de la réparation de l’ADN. (lien)
- Jacques Lebreton (Université de Nantes), CEISAM, équipe SYMBIOSE.
- Bruno Linclau (Université de Southampton) (lien)
- Steeve Thany (Université d’Orléans), Laboratoire de Biologie des Ligneux et des Grandes Cultures, UPRES EA 1207 (lien)
- Daniel Vercauteren & Laurence Leherte (université de Namur) Unité de chimie physique théorique et structurale, Laboratoire de chimie théorique (lien)